RNASeq数据下载处理全流程
最近需要重新跑一个物种的RNASeq数据处理,所以趁此机会记录一下。ssh win10@pcrent365.com -A 22522
一、下载数据
首先在NCBI官网上选择SRA数据库,输入想要的数据关键词搜索“Zea mays drought”
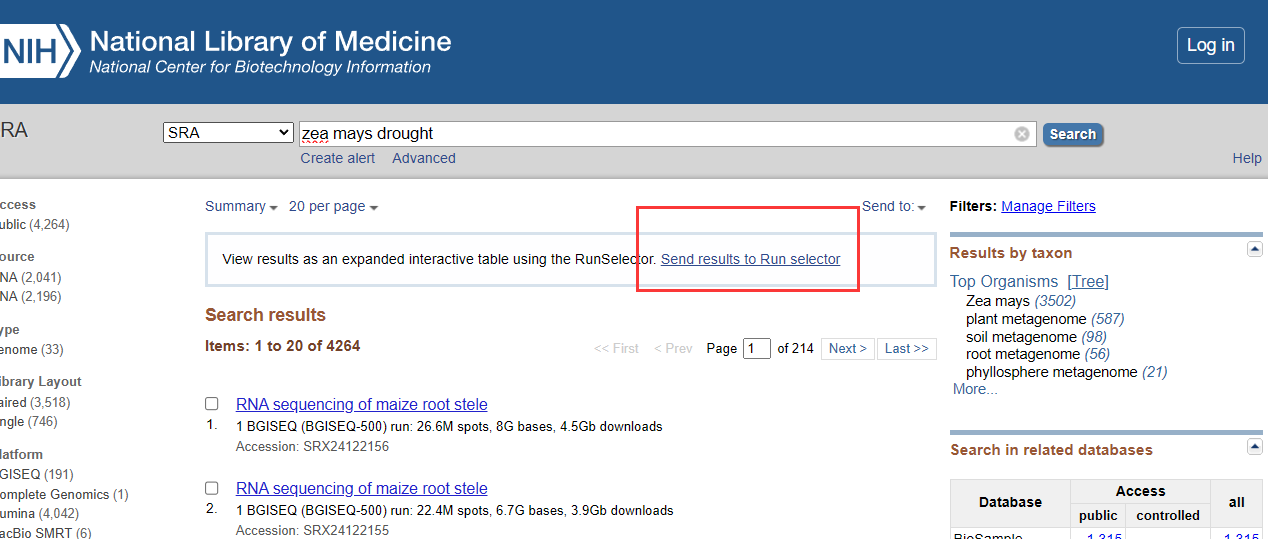
点击发送结果到数据框,点击Metadata下载SraRunTable。
接着对数据条进行筛选,可手动可写代码,看个人需求。筛选 ‘Assay Type’ 列为 ‘RNA Seq’ 的行和‘LibraryLayout’为‘PAIRED’
import pandas as pd |
筛选完之后对srr_id生成数据下载地址
import os |
windows的话得到下载链接之后复制到迅雷下载,一次复制一千条,正常在几十Mb/s
linux下采用多线程下载,正常在400Mb/s(看网络情况),存储够的话最好是提前下载
aria2c -i "/home/win/4T/GeneDL/RNASeq/SRR/zea_mays_download_urls_sra.txt" -x 16 |
下载也可参考第四步在全流程里面一起做
二、参考基因组索引 建立
首先在ensembl plants数据库下载对应物种的参考基因组序列.fa和注释文件.gff3,利用hisat进行索引建立
hisat2-build -p 4 "/home/win/4T/GeneDL/RNASeq/Reference/Zea_mays.fa" Zea_mays |
比对完之后,将.ht2文件放在一个文件夹Zea_mays下,后续比对需要用到这个文件夹
mv *.ht2 /home/win/4T/GeneDL/RNASeq/Reference/Zea_mays/ |
三、数据全流程自动处理
这里将数据下载,解压 ,比对,定量的步骤采用自动化的方式处理,需提前搭建包含parallel-fastq-dump、hisat2、samtools、featureCounts等软件。
环境配置
conda create -n rnaseq python=3.7 |
采用shell自动化命令
nj=8 #多少个进程 |
里面的py文件内容如下
# import sys |
发现问题停止运行的脚本stop.sh,如果停止运行了,要把srr,fastq,sam,bam中间文件删除,以免下一次报错
pkill -f "python3 /home/win/16t2/study/deep_learning/gene/course_try/1Sequence/RNASeq_allprocess.py" |
四、定量文件拼接与数据标准化
将定量出的tsv文件进行拼接以及转为FPKM
|
五、将数据从一个服务器传到另一个服务器
#进 在本地服务器运行 前面是本地服务器地址,后面是远端服务器 出---相对于本地 |
六、差异表达、富集分析、WGCNA分析
差异分析代码
import omicverse as ov |
富集分析代码GO
import gseapy as gp |
富集分析代码KEGG
import gseapy as gp |
WGCNA分析R
#Rscript script_name.R arg1 arg2 |
input.json如下
{"exp_path":"/home/win/4T/GeneDL/OSDrought_GCNAT_Link/plot/multispecies_WGCNA/indica/data/indica_CD_WGCNA.txt","geneMode":"mad","geneModeValue":"50","sampleCut":"1","netType":"unsigned","oldFolder":"","minModuleSize":"30","deepSplit":"3","mergeCutHeight":"0.25","method":"module"} |
后续就可以用数据做深度学习模型了~